Developing Body-Components-Based Theranostic Nanoparticles for Targeting Ovarian Cancer


 ,
,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparation of Fluorescein Isothiocyanate-Hyaluronic Acid-Bovine Serum Albumin (FITC-HA-BSA) Conjugates
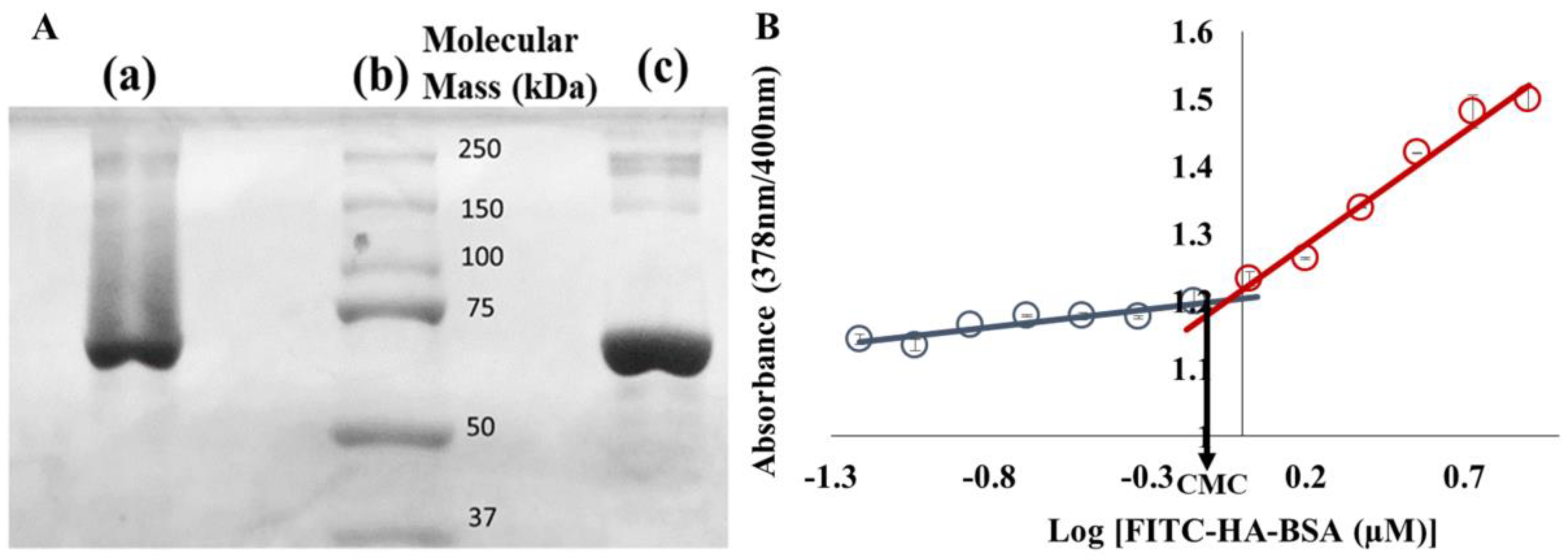
2.2.2. Determination of the Critical Micellization Concentration (CMC)
2.2.3. Preparation of FITC-HA-BSA–Paclitaxel Nanoparticles (PTX NPs)
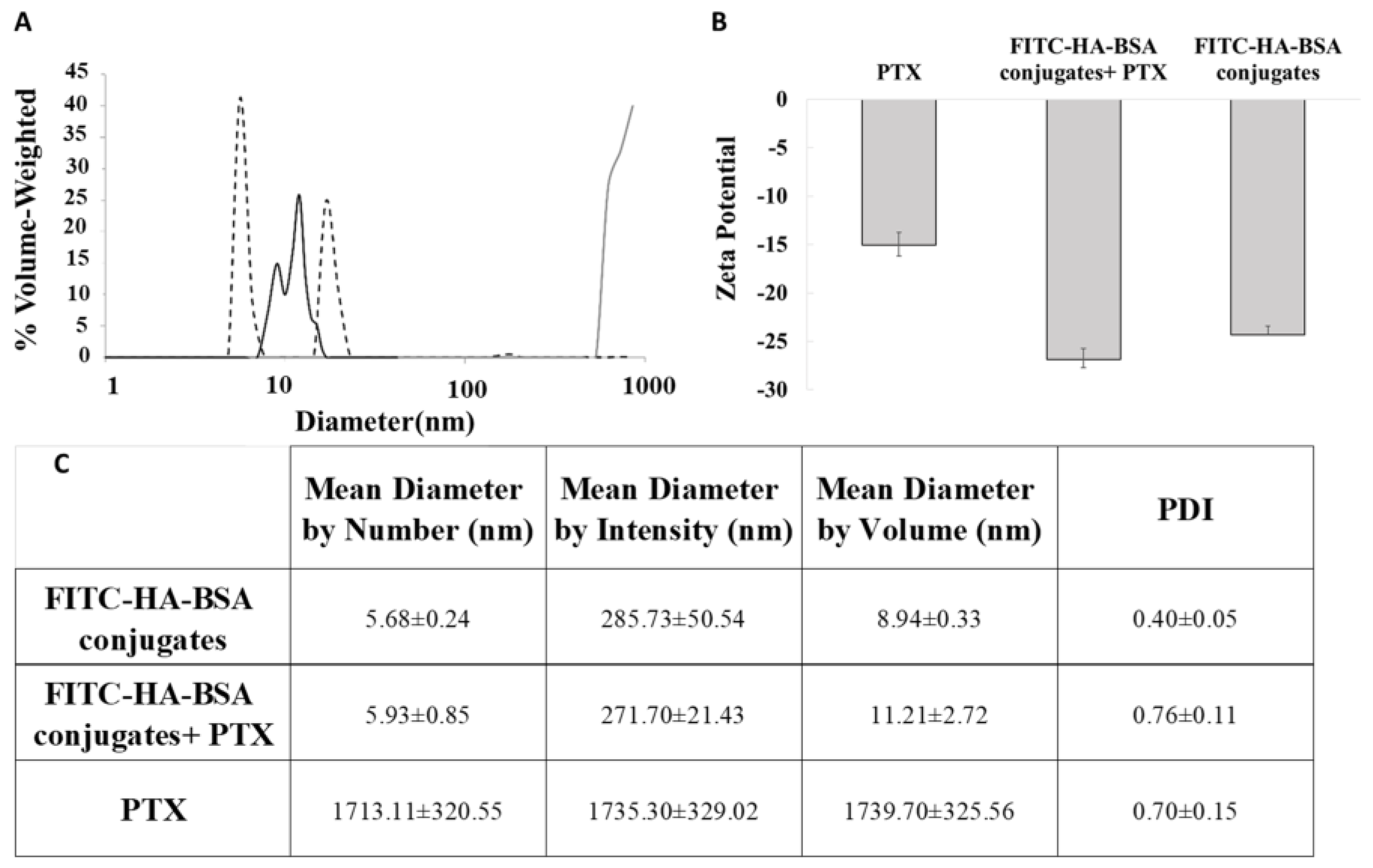
2.2.4. Size Determination
2.2.5. Zeta Potential
2.2.6. Light Microscopy
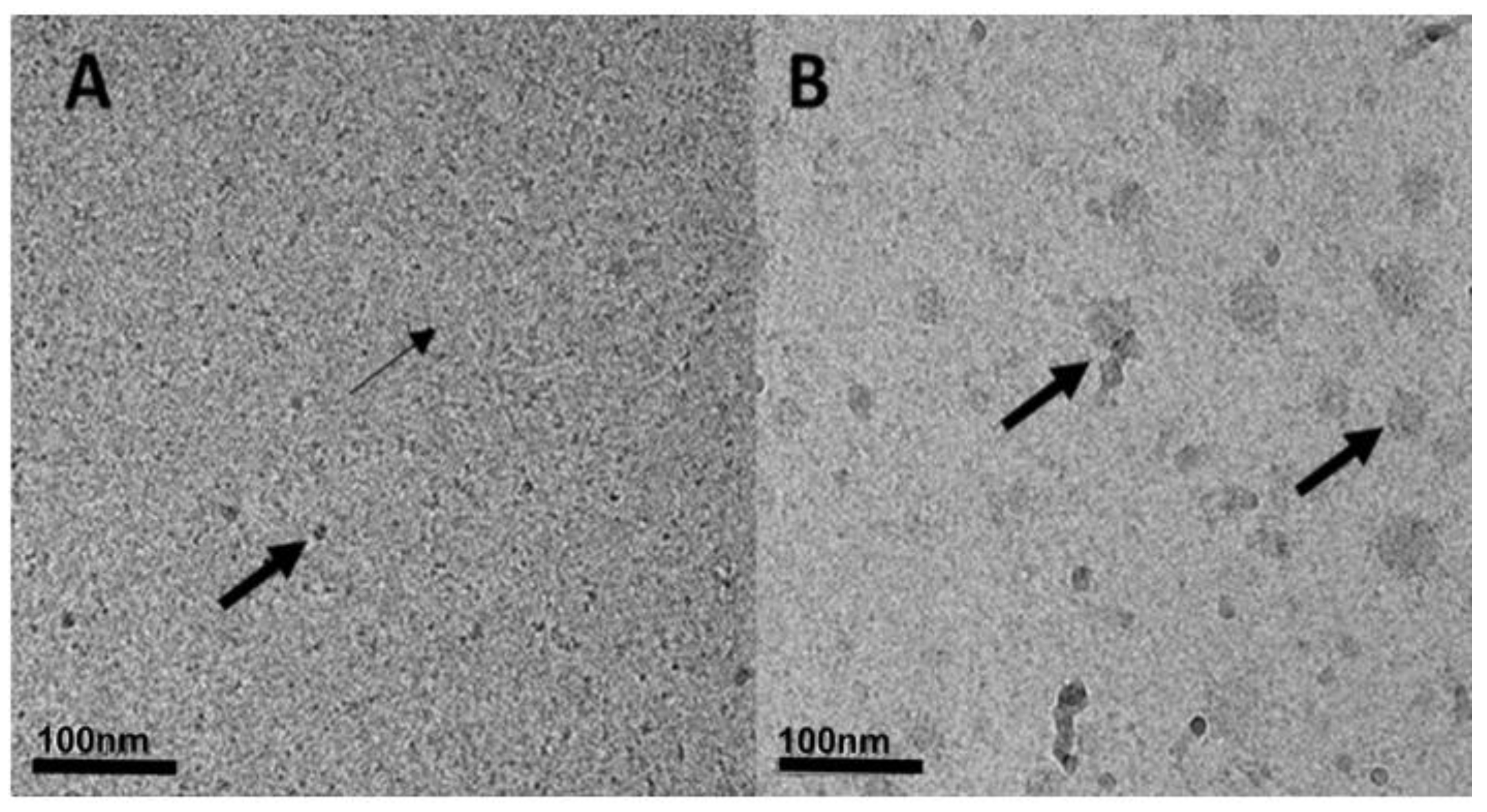
2.2.7. Cryogenic-Transmission Electron Microscopy (Cryo-TEM) Imaging
2.2.8. Encapsulation Efficiency (EE)
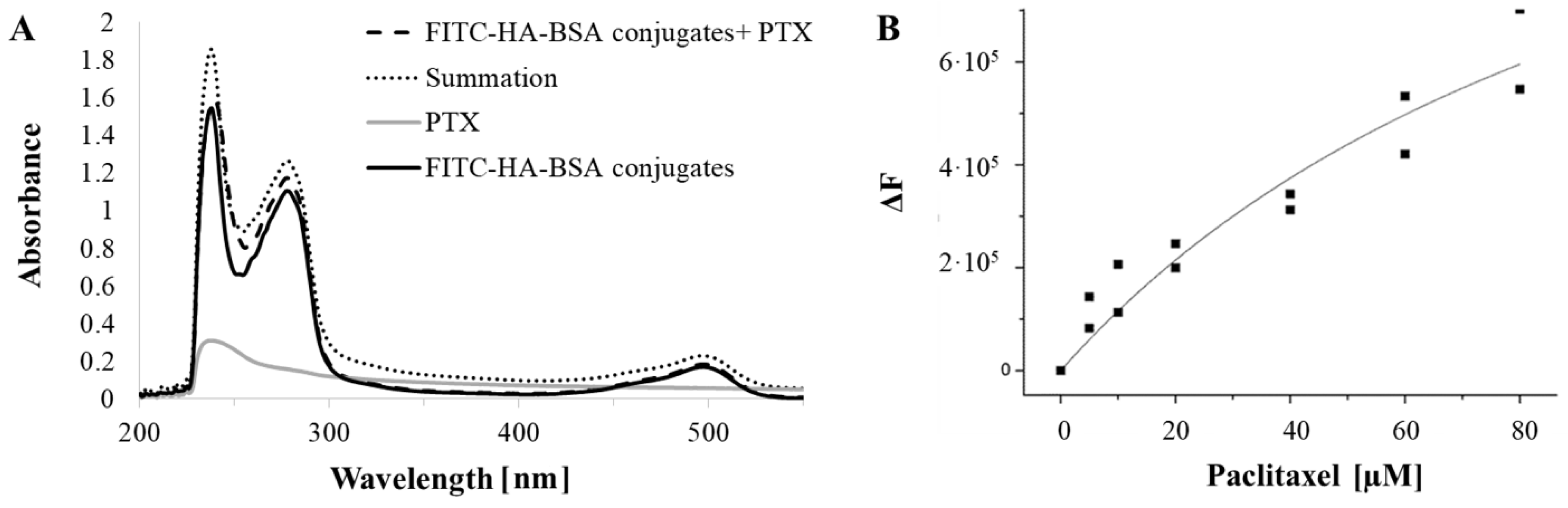
2.2.9. Binding Studies by Spectrophotometry
2.2.10. Binding Studies by Spectrofluorometry
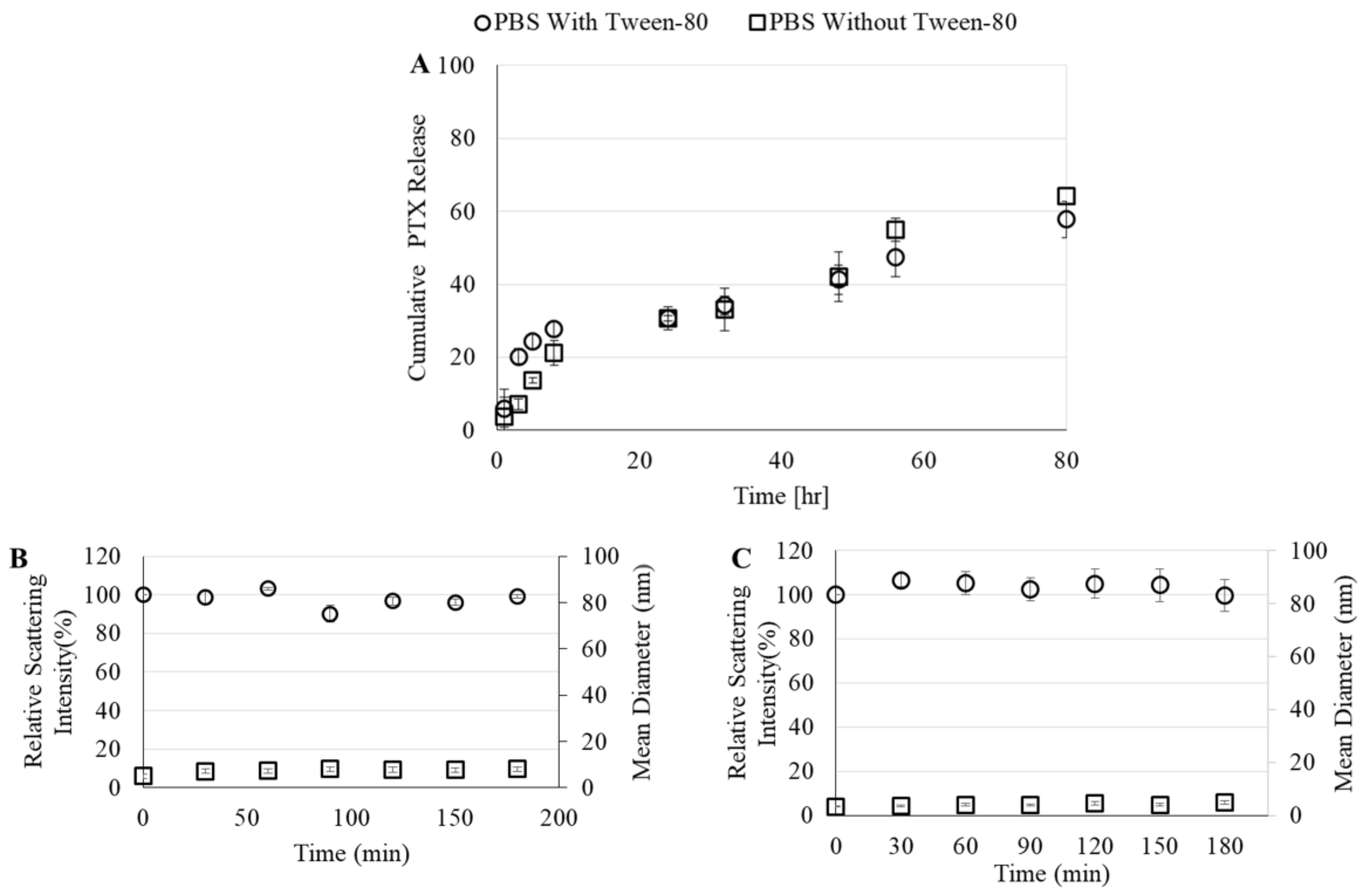
2.2.11. In Vitro PTX Release
2.2.12. Stability of FITC-HA-BSA Conjugates
2.2.13. Tissue Culture
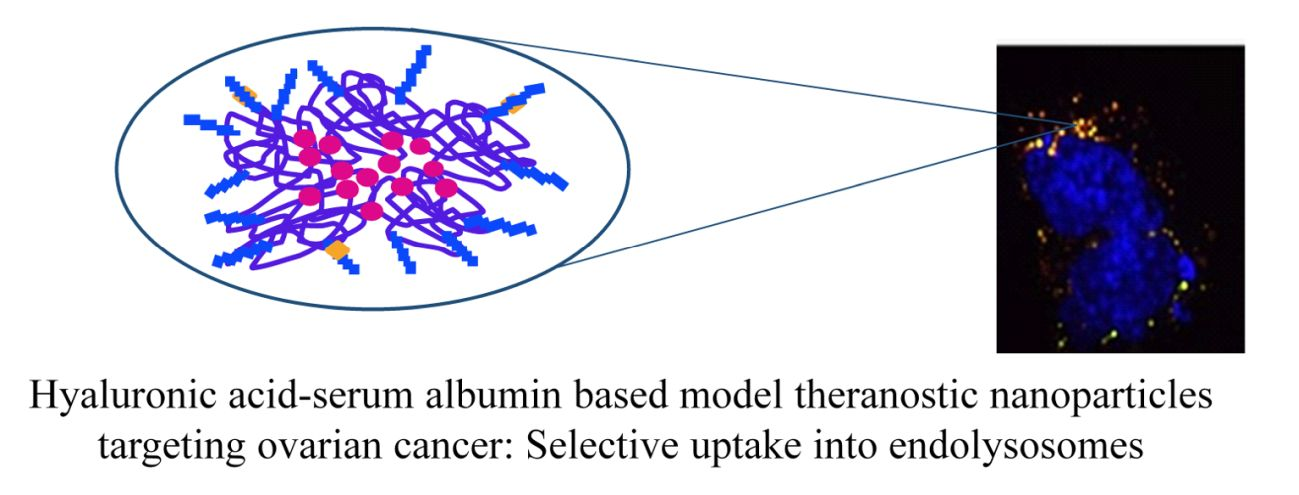
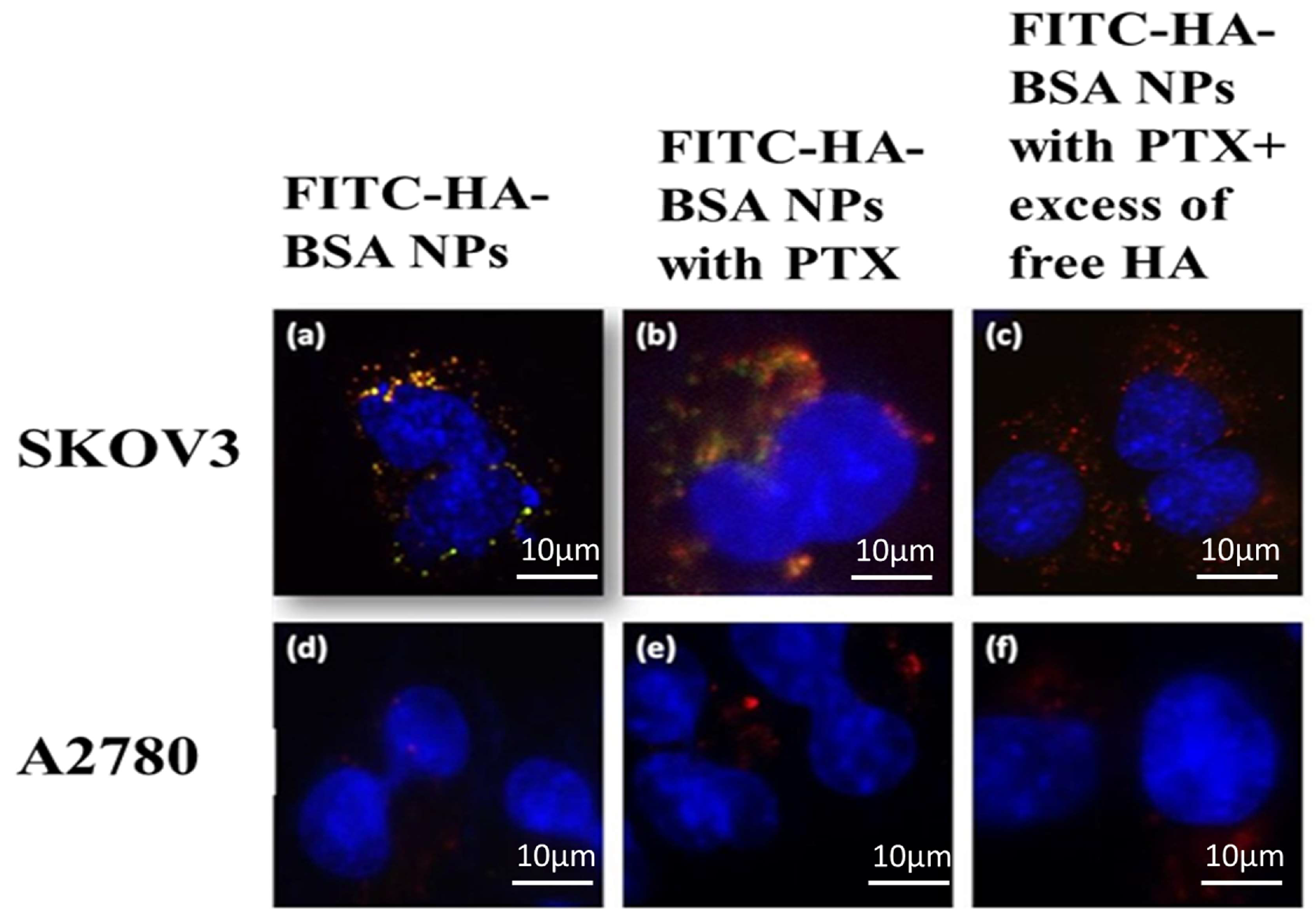
2.2.14. Selective Targeting
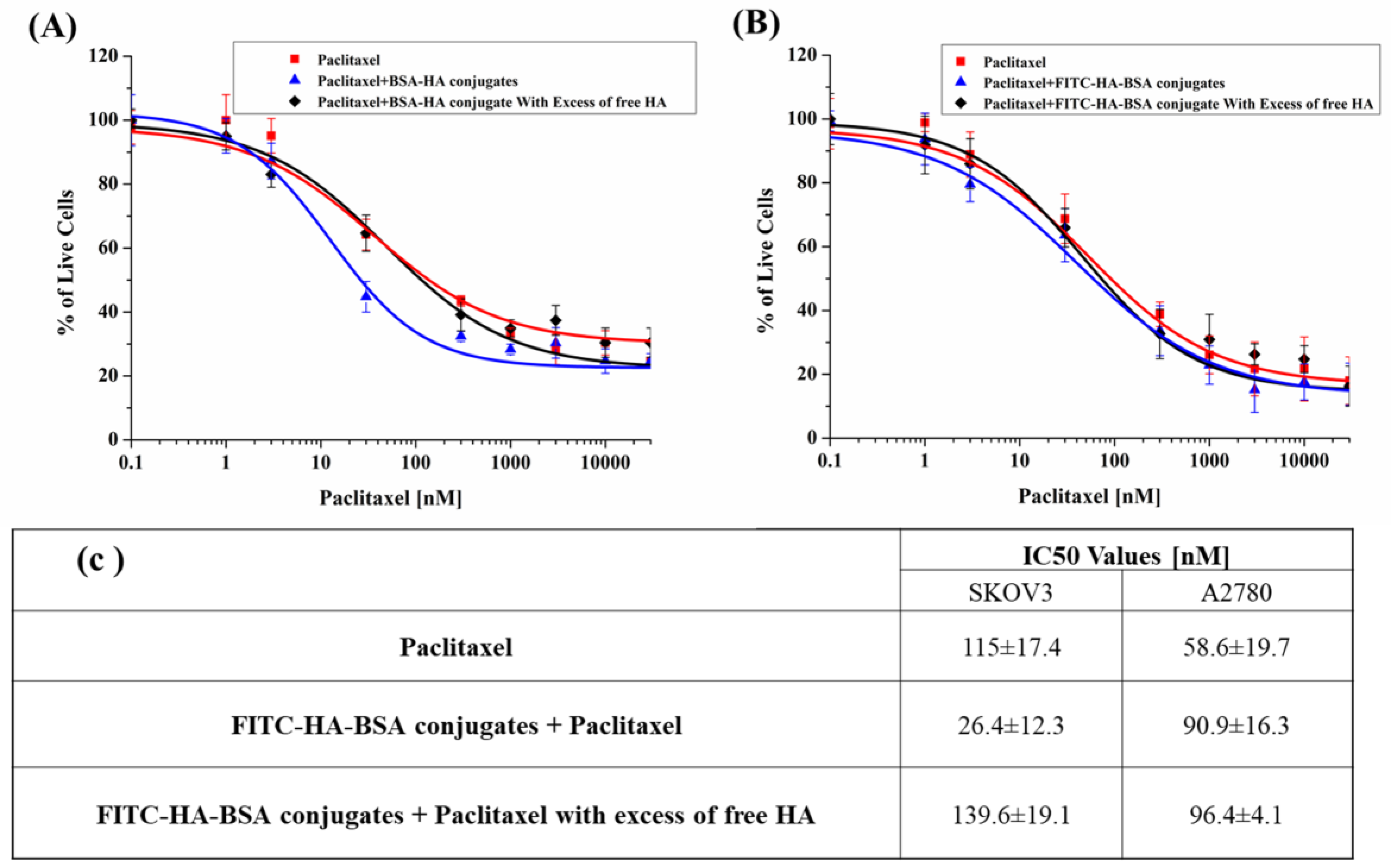
2.2.15. Cytotoxicity Assays
2.2.16. Statistical Analysis
3. Results
3.1. Characterization of FITC-HA-BSA Conjugates
Conjugation of FITC-HA to BSA
3.2. Encapsulation of PTX within FITC-HA-BSA Conjugates
3.2.1. Association Constant Determination
3.2.2. Particle Size Determination
3.2.3. Drug-Loading Capacity (LC) and Encapsulation Efficiency (EE)
3.2.4. PTX Release
3.2.5. Selective Targeting to CD44-Overexpressing Tumor Cells
3.2.6. PTX Cytotoxicity
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics. Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Livney, Y.D.; Assaraf, Y.G. Rationally designed nanovehicles to overcome cancer chemoresistance. Adv. Drug Deliv. Rev. 2013, 65, 1716–1730. [Google Scholar] [CrossRef] [PubMed]
- Shapira, A.; Livney, Y.D.; Broxterman, H.J.; Assaraf, Y.G. Nanomedicine for targeted cancer therapy: Towards the overcoming of drug resistance. Drug Resist. Updat. 2011, 14, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Zhitomirsky, B.; Yunaev, A.; Kreiserman, R.; Kaplan, A.; Stark, M.; Assaraf, Y.G. Lysosomotropic drugs activate TFEB via lysosomal membrane fluidization and consequent inhibition of mTORC1 activity. Cell Death Dis. 2018, 9, 1191. [Google Scholar] [CrossRef] [PubMed]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal accumulation of anticancer drugs triggers lysosomal exocytosis. Oncotarget 2017, 8, 45117–45132. [Google Scholar] [CrossRef]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomes as mediators of drug resistance in cancer. Drug Resist. Updat. 2016, 24, 23–33. [Google Scholar] [CrossRef]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal sequestration of hydrophobic weak base chemotherapeutics triggers lysosomal biogenesis and lysosome-dependent cancer multidrug resistance. Oncotarget 2015, 6, 1143–1156. [Google Scholar] [CrossRef]
- Khosroushahi, A.; Naderi-Manesh, H.; Yeganeh, H.; Barar, J.; Omidi, Y. Novel water-soluble polyurethane nanomicelles for cancer chemotherapy: Physicochemical characterization and cellular activities. J. Nanobiotechnol. 2012, 10, 2. [Google Scholar] [CrossRef]
- Hyung, W.; Ko, H.; Park, J.; Lim, E.; Park, S.B.; Park, Y.-J.; Yoon, H.G.; Suh, J.S.; Haam, S.; Huh, Y.-M. Novel hyaluronic acid (HA) coated drug carriers (HCDCs) for human breast cancer treatment. Biotechnol. Bioeng. 2008, 99, 442–454. [Google Scholar] [CrossRef]
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A Phase 3 Trial of Bevacizumab in Ovarian Cancer. N. Engl. J. Med. 2011, 365, 2484–2496. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Mumper, R.J. Nanomedicinal strategies to treat multidrug-resistant tumors: Current progress. Nanomedicine 2010, 5, 597–615. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; Cibula, D.; Benzaquen, A.O.; Poole, C.; Mathijssen, R.H.; Sonke, G.S.; Colombo, N.; Špaček, J.; Vuylsteke, P.; Hirte, H.; et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: A randomised phase 2 trial. Lancet Oncol. 2015, 16, 87–97. [Google Scholar] [CrossRef]
- Engelberg, S.; Modrejewski, J.; Walter, J.G.; Livney, Y.D.; Assaraf, Y.G. Cancer cell-selective, clathrin-mediated endocytosis of aptamerdecorated nanoparticles. Oncotarget 2018, 9, 20993–21006. [Google Scholar] [CrossRef] [PubMed]
- Han, H.S.; Choi, K.Y.; Ko, H.; Jeon, J.; Saravanakumar, G.; Suh, Y.D.; Lee, D.S.; Park, J.H. Bioreducible core-crosslinked hyaluronic acid micelle for targeted cancer therapy. J. Control. Release 2015, 200, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lan, C.Q.; Post, M.; Simard, B.; Deslands, Y.; Hsieh, T.H. Design of nanoparticles as drug carriers for cancer therapy. Cancer Genom. Proteom. 2006, 3, 147–157. [Google Scholar]
- Zhao, H.; Yung, L.Y.L. Selectivity of folate conjugated polymer micelles against different tumor cells. Int. J. Pharm. 2008, 349, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Kun, R.; Kis, L.; Dékány, I. Hydrophobization of bovine serum albumin with cationic surfactants with different hydrophobic chain length. Colloids Surf. B Biointerfaces 2010, 79, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Montagner, I.M.; Banzato, A.; Zuccolotto, G.; Renier, D.; Campisi, M.; Bassi, P.; Zanovello, P.; Rosato, A. Paclitaxel-hyaluronan hydrosoluble bioconjugate: Mechanism of action in human bladder cancer cell lines. Urol Oncol. 2013, 31, 1261–1269. [Google Scholar] [CrossRef]
- Yadav, A.K.; Mishra, P.; Agrawal, G.P. An insight on hyaluronic acid in drug targeting and drug delivery. J. Drug Target. 2008, 16, 91–107. [Google Scholar] [CrossRef]
- Bae, K.H.; Tan, S.; Yamashita, A.; Ang, W.X.; Gao, S.J.; Wang, S.; Chung, J.E.; Kurisawa, M. Hyaluronic acid-green tea catechin micellar nanocomplexes: Fail-safe cisplatin nanomedicine for the treatment of ovarian cancer without off-target toxicity. Biomaterials 2017, 148, 41–53. [Google Scholar] [CrossRef]
- Yang, Y.; Jing, L.; Li, X.; Lin, L.; Yue, X.; Dai, Z. Hyaluronic Acid Conjugated Magnetic Prussian Blue@Quantum Dot Nanoparticles for Cancer Theranostics. Theranostics 2017, 7, 466–481. [Google Scholar] [CrossRef]
- Jin, Y.; Ma, X.; Feng, S.; Liang, X.; Dai, Z.; Tian, J.; Yue, X. Hyaluronic Acid Modified Tantalum Oxide Nanoparticles Conjugating Doxorubicin for Targeted Cancer Theranostics. Bioconjug. Chem. 2015, 26, 2530–2541. [Google Scholar] [CrossRef]
- Cho, H.J.; Yoon, I.S.; Yoon, H.Y.; Koo, H.; Jin, Y.J.; Ko, S.H.; Shim, J.S.; Kim, K.; Kwon, I.C.; Kim, D.D. Polyethylene glycol-conjugated hyaluronic acid-ceramide self-assembled nanoparticles for targeted delivery of doxorubicin. Biomaterials 2012, 33, 1190–1200. [Google Scholar] [CrossRef]
- Cho, E.J.; Sun, B.; Doh, K.O.; Wilson, E.M.; Torregrosa-Allen, S.; Elzey, B.D.; Yeo, Y. Intraperitoneal delivery of platinum with in-situ crosslinkable hyaluronic acid gel for local therapy of ovarian cancer. Biomaterials 2015, 37, 312–319. [Google Scholar] [CrossRef]
- Homma, A.; Sato, H.; Tamura, T.; Okamachi, A.; Emura, T.; Ishizawa, T.; Kato, T.; Matsuura, T.; Sato, S.; Higuchi, Y.; et al. Synthesis and optimization of hyaluronic acid–methotrexate conjugates to maximize benefit in the treatment of osteoarthritis. Bioorganic Med. Chem. 2010, 18, 1062–1075. [Google Scholar] [CrossRef]
- Ravindran, A.; Singh, A.; Raichur, A.M.; Chandrasekaran, N.; Mukherjee, A. Studies on interaction of colloidal Ag nanoparticles with bovine serum albumin (BSA). Colloids Surf. B Biointerfaces 2010, 76, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Jeyachandran, Y.L.; Mielczarski, E.; Rai, B.; Mielczarski, J.A. Quantitative and Qualitative Evaluation of Adsorption/Desorption of Bovine Serum Albumin on Hydrophilic and Hydrophobic Surfaces. Langmuir 2009, 25, 11614–11620. [Google Scholar] [CrossRef]
- Lomis, N.; Westfall, S.; Farahdel, L.; Malhotra, M.; Shum-Tim, D.; Prakash, S. Human Serum Albumin Nanoparticles for Use in Cancer Drug Delivery: Process Optimization and In Vitro Characterization. Nanomaterials 2016, 6, 116. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, M.; Rajabi, M.; Mousa, S.; Srinivasan, M.; Rajabi, M.; Mousa, S.A. Multifunctional Nanomaterials and Their Applications in Drug Delivery and Cancer Therapy. Nanomaterials 2015, 5, 1690–1703. [Google Scholar] [CrossRef] [PubMed]
- Raju, G.; Benton, L.; Pavitra, E.; Yu, J.S. Multifunctional nanoparticles: Recent progress in cancer therapeutics. Chem. Commun. 2015, 51, 13248–13529. [Google Scholar] [CrossRef] [PubMed]
- Low, P.S.; Henne, W.A.; Doorneweerd, D.D. Discovery and Development of Folic-Acid-Based Receptor Targeting for Imaging and Therapy of Cancer and Inflammatory Diseases. Acc. Chem. Res. 2008, 41, 120–129. [Google Scholar] [CrossRef]
- Pinhassi, R.I.; Assaraf, Y.G.; Farber, S.; Stark, M.; Ickowicz, D.; Drori, S.; Domb, A.J.; Livney, Y.D. Arabinogalactan−Folic Acid−Drug Conjugate for Targeted Delivery and Target-Activated Release of Anticancer Drugs to Folate Receptor-Overexpressing Cells. Biomacromolecules 2010, 11, 294–303. [Google Scholar] [CrossRef]
- Chen, B.; Miller, R.J.; Dhal, P.K. Hyaluronic acid-based drug conjugates: State-of-the-art and perspectives. J. Biomed. Nanotechnol. 2014, 10, 4–16. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, C.; Cheng, L.; He, W.; Cheng, Z.; Liu, Z. Protein modified upconversion nanoparticles for imaging-guided combined photothermal and photodynamic therapy. Biomaterials 2014, 35, 2915–2923. [Google Scholar] [CrossRef]
- Edelman, R.; Assaraf, Y.G.; Levitzky, I.; Shahar, T.; Livney, Y.D. Hyaluronic acid-serum albumin conjugate-based nanoparticles for targeted cancer therapy. Oncotarget 2017, 8, 24337–24353. [Google Scholar] [CrossRef] [PubMed]
- Luk, B.T.; Fang, R.H.; Zhang, L. Lipid-and polymer-based nanostructures for cancer theranostics. Theranostics 2012, 2, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Liu, J.; Gu, X.; Gong, H.; Shi, X.; Liu, T.; Wang, C.; Wang, X.; Liu, G.; Xing, H.; et al. PEGylated WS 2 Nanosheets as a Multifunctional Theranostic Agent for in vivo Dual-Modal CT/Photoacoustic Imaging Guided Photothermal Therapy. Adv. Mater. 2014, 26, 1886–1893. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, J.; Nie, X.; Wen, T.; Ji, Y.; Wu, X.; Zhao, Y.; Chen, C. Near Infrared Laser-Induced Targeted Cancer Therapy Using Thermoresponsive Polymer Encapsulated Gold Nanorods. J. Am. Chem. Soc. 2014, 136, 7317–7326. [Google Scholar] [CrossRef]
- Oliveira, H.; Pérez-Andrés, E.; Thevenot, J.; Sandre, O.; Berra, E.; Lecommandoux, S. Magnetic field triggered drug release from polymersomes for cancer therapeutics. J. Controlled Release 2013, 169, 165–170. [Google Scholar] [CrossRef]
- Shi, X.; Gong, H.; Li, Y.; Wang, C.; Cheng, L.; Liu, Z. Graphene-based magnetic plasmonic nanocomposite for dual bioimaging and photothermal therapy. Biomaterials 2013, 34, 4786–4793. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Liu, D.; Song, S.; Wang, X.; Zhang, H. Graphene oxide covalently grafted upconversion nanoparticles for combined NIR mediated imaging and photothermal/photodynamic cancer therapy. Biomaterials 2013, 34, 7715–7724. [Google Scholar] [CrossRef]
- Gao, F.P.; Lin, Y.X.; Li, L.L.; Liu, Y.; Mayerhöffer, U.; Spenst, P.; Su, J.G.; Li, J.Y.; Würthner, F.; Wang, H. Supramolecular adducts of squaraine and protein for noninvasive tumor imaging and photothermal therapy in vivo. Biomaterials 2014, 35, 1004–1014. [Google Scholar] [CrossRef]
- Wang, C.; Ravi, S.; Garapati, U.S.; Das, M.; Howell, M.; MallelaMallela, J.; Alwarapan, S.; Mohapatra, S.S.; Mohapatra, S. Multifunctional chitosan magnetic-graphene (CMG) nanoparticles: A theranostic platform for tumor-targeted co-delivery of drugs, genes and MRI contrast agents. J. Mater. Chem. B 2013, 1, 4396–4405. [Google Scholar] [CrossRef]
- Petersen, A.L.; Hansen, A.E.; Gabizon, A.; Andresen, T.L. Liposome imaging agents in personalized medicine. Adv. Drug Deliv. Rev. 2012, 64, 1417–1435. [Google Scholar] [CrossRef]
- Brakmane, G.; Madani, S.Y.; Seifalian, A. Cancer Antibody Enhanced Real Time Imaging Cell Probes–a Novel Theranostic Tool using Polymer Linked Carbon Nanotubes and Quantum Dots. Anti-Cancer Agents Med. Chem. 2013, 13, 821–832. [Google Scholar] [CrossRef]
- Wegner, K.D.; Hildebrandt, N. Quantum dots: Bright and versatile in vitro and in vivo fluorescence imaging biosensors. Chem. Soc. Rev. 2015, 44, 4792–4843. [Google Scholar] [CrossRef]
- Wang, Y.; Black, K.C.; Luehmann, H.; Li, W.; Zhang, Y.; Cai, X.; Wan, D.; Liu, S.Y.; Li, M.; Kim, P.; et al. Comparison Study of Gold Nanohexapods, Nanorods, and Nanocages for Photothermal Cancer Treatment. ACS Nano 2013, 7, 2068–2077. [Google Scholar] [CrossRef]
- Mieszawska, A.J.; Mulder, W.J.M.; Fayad, Z.A.; Cormode, D.P. Multifunctional Gold Nanoparticles for Diagnosis and Therapy of Disease. Mol. Pharm. 2013, 10, 831–847. [Google Scholar] [CrossRef]
- Wang, C.; Xu, H.; Liang, C.; Liu, Y.; Li, Z.; Yang, G.; Cheng, L.; Li, Y.; Liu, Z. Iron Oxide @ Polypyrrole Nanoparticles as a Multifunctional Drug Carrier for Remotely Controlled Cancer Therapy with Synergistic Antitumor Effect. ACS Nano 2013, 7, 6782–6795. [Google Scholar] [CrossRef]
- Yang, H.; Mao, H.; Wan, Z.; Zhu, A.; Guo, M.; Li, Y.; Li, X.; Wan, J.; Yang, X.; Shuai, X.; et al. Micelles assembled with carbocyanine dyes for theranostic near-infrared fluorescent cancer imaging and photothermal therapy. Biomaterials 2013, 34, 9124–9133. [Google Scholar] [CrossRef]
- Muthu, M.S.; Kutty, R.V.; Luo, Z.; Xie, J.; Feng, S.S. Theranostic vitamin E TPGS micelles of transferrin conjugation for targeted co-delivery of docetaxel and ultra bright gold nanoclusters. Biomaterials 2015, 39, 234–248. [Google Scholar] [CrossRef]
- Li, S.; Goins, B.; Zhang, L.; Bao, A. Novel Multifunctional Theranostic Liposome Drug Delivery System: Construction, Characterization, and Multimodality MR, Near-Infrared Fluorescent, and Nuclear Imaging. Bioconjug. Chem. 2012, 23, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Al-Jamal, W.T.; Kostarelos, K. Liposomes: From a Clinically Established Drug Delivery System to a Nanoparticle Platform for Theranostic Nanomedicine. Acc. Chem. Res. 2011, 44, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Kannan, R.M.; Nance, E.; Kannan, S.; Tomalia, D.A. Emerging concepts in dendrimer-based nanomedicine: From design principles to clinical applications. J. Intern. Med. 2014, 276, 579–617. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Datir, S.R.; Singh, R.P.; Jain, S. Augmented Anticancer Activity of a Targeted, Intracellularly Activatable, Theranostic Nanomedicine Based on Fluorescent and Radiolabeled, Methotrexate-Folic Acid-Multiwalled Carbon Nanotube Conjugate. Mol. Pharm. 2013, 10, 2543–2557. [Google Scholar] [CrossRef] [PubMed]
- De Volder, M.F.; Tawfick, S.H.; Baughman, R.H.; Hart, A.J. Carbon nanotubes: Present and future commercial applications. Science 2013, 339, 535–539. [Google Scholar] [CrossRef]
- Ye, S.; Marston, G.; McLaughlan, J.R.; Sigle, D.O.; Ingram, N.; Baumberg, S.F.J.J.; Bushby, R.J.; Markham, A.F.; Critchley, K.; Coletta, P.L.; et al. Engineering Gold Nanotubes with Controlled Length and Near-Infrared Absorption for Theranostic Applications. Adv. Funct. Mater. 2015, 25, 2117–2127. [Google Scholar] [CrossRef]
- Chen, Q.; Liang, C.; Wang, X.; He, J.; Li, Y.; Liu, Z. An albumin-based theranostic nano-agent for dual-modal imaging guided photothermal therapy to inhibit lymphatic metastasis of cancer post surgery. Biomaterials 2014, 35, 9355–9362. [Google Scholar] [CrossRef]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef]
- Gonen, N.; Assaraf, Y.G. Antifolates in cancer therapy: Structure, activity and mechanisms of drug resistance. Drug Resist. Updat. 2012, 15, 183–210. [Google Scholar] [CrossRef]
- Drori, S.; Jansen, G.; Mauritz, R.; Peters, G.J.; Assaraf, Y.G. Clustering of mutations in the first transmembrane domain of the human reduced folate carrier in GW1843U89-resistant leukemia cells with impaired antifolate transport and augmented folate uptake. J. Biol. Chem. 2000, 275, 30855–30863. [Google Scholar] [CrossRef]
- Kaufman, Y.; Ifergan, I.; Rothem, L.; Jansen, G.; Assaraf, Y.G. Coexistence of multiple mechanisms of PT523 resistance in human leukemia cells harboring 3 reduced folate carrier alleles: Transcriptional silencing, inactivating mutations, and allele loss. Blood 2006, 107, 3288–3294. [Google Scholar] [CrossRef][Green Version]
- El-Dakdouki, M.H.; El-Boubbou, K.; Zhu, D.C.; Huang, X. A simple method for the synthesis of hyaluronic acid coated magnetic nanoparticles for highly efficient cell labelling and in vivo imaging. RSC Adv. 2011, 1, 1449. [Google Scholar] [CrossRef]
- Zhang, L.; He, Y.; Ma, G.; Song, C.; Sun, H. Paclitaxel-loaded polymeric micelles based on poly (ε-caprolactone)-poly (ethylene glycol)-poly (ε-caprolactone) triblock copolymers: In vitro and in vivo. Nanomedicine 2012, 8, 925–934. [Google Scholar] [CrossRef]
- Bar-Zeev, M.; Kelmansky, D.; Assaraf, Y.G.; Livney, Y.D. β-Casein micelles for oral delivery of SN-38 and elacridar to overcome BCRP-mediated multidrug resistance in gastric cancer. Eur. J. Pharm. Biopharm. 2018, 133, 240–249. [Google Scholar] [CrossRef]
- Koo, A.N.; Min, K.H.; Lee, H.J.; Lee, S.U.; Kim, K.; Kwon, I.C.; Cho, S.H.; Jeong, S.Y.; Lee, S.C. Tumor accumulation and antitumor efficacy of docetaxel-loaded core-shell-corona micelles with shell-specific redox-responsive cross-links. Biomaterials 2012, 33, 1489–1499. [Google Scholar] [CrossRef]
- Amann, E.; Wolff, P.; Breel, E.; van Griensven, M.; Balmayor, E.R. Hyaluronic acid facilitates chondrogenesis and matrix deposition of human adipose derived mesenchymal stem cells and human chondrocytes co-cultures. Acta Biomater. 2017, 52, 130–144. [Google Scholar] [CrossRef]
- Chattopadhyay, A.; London, E. Fluorimetric Determination of Critical Micelle Concentration avoiding interference from detergent charge. Aalytical Biochemistry. 1984, 139, 408–412. [Google Scholar] [CrossRef]
- Maruyama, K. Intracellular targeting delivery of liposomal drugs to solid tumors based on EPR effects. Adv. Drug Deliv. Rev. 2011, 63, 161–169. [Google Scholar] [CrossRef]
- Landesman-Milo, D.; Ramishetti, S.; Peer, D. Nanomedicine as an emerging platform for metastatic lung cancer therapy. Cancer Metastasis Rev. 2015, 34, 291–301. [Google Scholar] [CrossRef]
- Janib, S.; Moses, A.; MacKay, J.A. Drug delivery reviews, and undefined 2010. Imaging and drug delivery using theranostic nanoparticles. Adv. Drug Deliv. Rev. 2010, 30, 1052–1063. [Google Scholar] [CrossRef]
- Surapaneni, M.S.; Das, S.K.; Das, N.G. Designing Paclitaxel drug delivery systems aimed at improved patient outcomes: Current status and challenges. ISRN Pharmacol. 2012, 2012, 623139. [Google Scholar] [CrossRef]
- Bar-Zeev, M.; Assaraf, Y.G.; Livney, Y.D. β-casein nanovehicles for oral delivery of chemotherapeutic drug combinations overcoming P-glycoprotein-mediated multidrug resistance in human gastric. Oncotarget 2016, 7, 23322–23334. [Google Scholar] [CrossRef]
- Lin, W.J.; Juang, L.W.; Lin, C.C. Stability and release performance of a series of pegylated copolymeric micelles. Pharm. Res. 2003, 20, 668–673. [Google Scholar] [CrossRef]
- Cho, Y.W.; Lee, J.; Lee, S.C.; Huh, K.M.; Park, K. Hydrotropic agents for study of in vitro paclitaxel release from polymeric micelles. J. Control Release 2004, 97, 249–257. [Google Scholar] [CrossRef]
- Shin, H.C.; Alani, A.W.; Rao, D.A.; Rockich, N.C.; Kwon, G.S. Multi-drug loaded polymeric micelles for simultaneous delivery of poorly soluble anticancer drugs. J Control Release 2009, 140, 294–300. [Google Scholar] [CrossRef]
- Bao, G.; Mitragotri, S.; Tong, S. Multifunctional Nanoparticles for Drug Delivery and Molecular Imaging. Annu. Rev. Biomed. Eng. 2013, 15, 253–282. [Google Scholar] [CrossRef]
- Jiang, W.; Kim, B.Y.; Rutka, J.T.; Chan, W.C. Nanoparticle-mediated cellular response is size-dependent. Nat. Nanotechnol. 2008, 3, 145–150. [Google Scholar] [CrossRef]
- Gaur, U.; Sahoo, S.K.; De, T.K.; Ghosh, P.C.; Maitra, A.; Ghosh, P.K. Biodistribution of fluoresceinated dextran using novel nanoparticles evading reticuloendothelial system. Int. J. Pharm. 2000, 202, 1–10. [Google Scholar] [CrossRef]
- Bukowska, B.; Marczak, A. Innovative therapy of ovarian cancer based on overexpression of CD44 receptor. Ginekol Pol. 2015, 86, 388–391. [Google Scholar] [CrossRef]
- Luo, Y.; Ziebell, M.R.; Prestwich, G.D. A Hyaluronic Acid−Taxol Antitumor Bioconjugate Targeted to Cancer Cells. Biomacromolecules 2000, 1, 208–218. [Google Scholar] [CrossRef]
- Serafino, A.; Zonfrillo, M.; Andreola, F.; Psaila, R.; Mercuri, L.; Moroni, N.; Renier, D.; Campisi, M.; Secchieri, C.; Pierimarchi, P. CD44-targeting for antitumor drug delivery: A new SN-38-hyaluronan bioconjugate for locoregional treatment of peritoneal carcinomatosis. Curr. Cancer Drug Targets 2011, 11, 572–585. [Google Scholar] [CrossRef]
- Quan, Y.H.; Kim, B.; Park, J.-H.; Choi, Y.; Choi, Y.H.; Kim, H.K. Highly sensitive and selective anticancer effect by conjugated HA-cisplatin in non-small cell lung cancer overexpressed with CD44. Exp. Lung Res. 2014, 40, 475–484. [Google Scholar] [CrossRef]
- Rosso, F.; Quagliariello, V.; Tortora, C.; di Lazzaro, A.; Barbarisi, A.; Iaffaioli, R.V. Cross-linked hyaluronic acid sub-micron particles: In vitro and in vivo biodistribution study in cancer xenograft model. J. Mater. Sci. Mater. Med. 2013, 24, 1473–1481. [Google Scholar] [CrossRef]
- Rivkin, I.; Cohen, K.; Koffler, J.; Melikhov, D.; Peer, D.; Margalit, R. Paclitaxel-clusters coated with hyaluronan as selective tumor-targeted nanovectors. Biomaterials 2010, 31, 7106–7114. [Google Scholar] [CrossRef]
- Journo-Gershfeld, G.; Kapp, D.; Shamay, Y.; Kopeček, J.; David, A. Hyaluronan Oligomers-HPMA Copolymer Conjugates for Targeting Paclitaxel to CD44-Overexpressing Ovarian Carcinoma. Pharm. Res. 2012, 29, 1121–1133. [Google Scholar] [CrossRef]
- Lennon, F.E.; Mirzapoiazova, T.; Mambetsariev, N.; Mambetsariev, B.; Salgia, R.; Singleton, P.A. Transactivation of the receptor-tyrosine kinase ephrin receptor A2 is required for the low molecular weight hyaluronan-mediated angiogenesis that is implicated in. J. Biol. Chem. 2014, 289, 24043–24058. [Google Scholar] [CrossRef]
- Lesley, J.; Hascall, V.C.; Tammi, M.; Hyman, R. Hyaluronan binding by cell surface CD44. J. Biol. Chem. 2000, 275, 26967–26975. [Google Scholar] [CrossRef]
- Plattt, V.M.; Szoka, F.C. Anticancer therapeutics: Targeting macromolecules and nanocarriers to hyaluronan or CD44, a hyaluronan receptor. Mol. Pharm. 2008, 5, 474–486. [Google Scholar] [CrossRef]
- Frederiks, C.N.; Lam, S.W.; Guchelaar, H.J.; Boven, E. Genetic polymorphisms and paclitaxel-or docetaxel-induced toxicities: A systematic review. Cancer Treat Rev. 2015, 41, 935–950. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Edelman, R.; Assaraf, Y.G.; Slavkin, A.; Dolev, T.; Shahar, T.; Livney, Y.D. Developing Body-Components-Based Theranostic Nanoparticles for Targeting Ovarian Cancer. Pharmaceutics 2019, 11, 216. https://doi.org/10.3390/pharmaceutics11050216
Edelman R, Assaraf YG, Slavkin A, Dolev T, Shahar T, Livney YD. Developing Body-Components-Based Theranostic Nanoparticles for Targeting Ovarian Cancer. Pharmaceutics. 2019; 11(5):216. https://doi.org/10.3390/pharmaceutics11050216
Chicago/Turabian StyleEdelman, Ravit, Yehuda G. Assaraf, Anton Slavkin, Tamar Dolev, Tal Shahar, and Yoav D. Livney. 2019. "Developing Body-Components-Based Theranostic Nanoparticles for Targeting Ovarian Cancer" Pharmaceutics 11, no. 5: 216. https://doi.org/10.3390/pharmaceutics11050216
APA StyleEdelman, R., Assaraf, Y. G., Slavkin, A., Dolev, T., Shahar, T., & Livney, Y. D. (2019). Developing Body-Components-Based Theranostic Nanoparticles for Targeting Ovarian Cancer. Pharmaceutics, 11(5), 216. https://doi.org/10.3390/pharmaceutics11050216